Hurlers syndrome is a rare, progressive genetic disorder that affects how the body processes complex molecules essential for normal cellular function. As a severe form of mucopolysaccharidosis type I (MPS I), it leads to systemic accumulation of glycosaminoglycans, gradually impairing multiple organs. The condition often presents early in life, making timely diagnosis critical. Recent advances in treatment and emerging gene therapy approaches are beginning to shift how this disease is managed and understood.
What is Hurlers Syndrome?
Hurlers syndrome, also referred to as Hurler disease, results from a deficiency of the enzyme alpha-L-iduronidase. This enzyme is responsible for breaking down specific complex sugars within lysosomes. When it is absent or dysfunctional, these substances accumulate in tissues, disrupting normal cellular processes.
Unlike localized disorders, Hurlers syndrome is systemic. It affects skeletal development, organ function, and neurological health simultaneously. This widespread involvement explains both the early onset and progressive nature of the condition.
Causes of Hurlers Syndrome
The underlying cause is a mutation in the IDUA gene, inherited in an autosomal recessive pattern. This means both parents must carry the defective gene for the child to be affected.
From a biological perspective, the mutation leads to enzyme deficiency, which in turn disrupts lysosomal degradation pathways. The result is progressive storage of undegraded material within cells, particularly in connective tissue, cartilage, and organs.
Symptoms of Hurlers Syndrome
Symptoms evolve over time, often beginning subtly and becoming more pronounced as accumulation increases.
In early stages, children may present with developmental delay, recurrent infections, and hernias. As the disease progresses, physical characteristics become more evident, including coarse facial features and joint stiffness. Organ enlargement, particularly of the liver and spleen, is also common.
Neurological involvement introduces another layer of complexity. Cognitive decline, behavioral changes, and reduced learning ability may develop, especially if treatment is delayed.
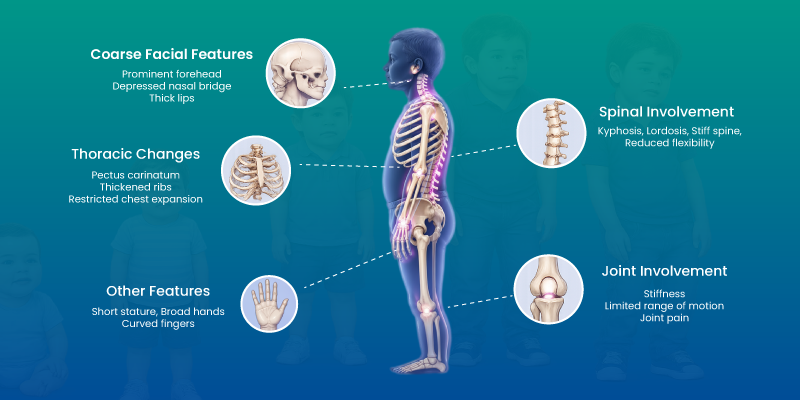
How Hurlers Syndrome Affects the Body
The disease does not affect a single system in isolation. Instead, it creates a cascade of dysfunction across multiple organ systems.
| System | Clinical Impact |
| Skeletal | Abnormal bone growth, joint restriction |
| Cardiac | Valve thickening, reduced function |
| Respiratory | Airway narrowing, recurrent infections |
| Neurological | Cognitive decline, developmental delay |
This multi-system involvement requires coordinated management across specialties rather than isolated treatment.
Diagnosis of Hurlers Syndrome
Prenatal Diagnosis
Prenatal detection is possible through genetic testing when there is known family history. Enzyme activity can also be assessed using specialized screening methods.
Early identification allows for planning of intervention strategies immediately after birth.

Postnatal Diagnosis
After birth, diagnosis is typically initiated based on clinical presentation. Confirmatory testing includes enzyme assays and genetic analysis.
A structured diagnostic pathway ensures differentiation from other lysosomal storage disorders, which may present with overlapping features.
Treatment of Hurlers Syndrome
Treatment approaches are layered, combining disease-modifying strategies with supportive care.
Disease-Modifying Treatments
Two primary interventions are currently used to alter disease progression.
Enzyme replacement therapy introduces the missing enzyme into the body, helping reduce substrate accumulation in peripheral tissues. However, its effect on neurological symptoms is limited.
Hematopoietic stem cell transplantation offers a more sustained source of enzyme production. When performed early, it can slow disease progression and improve long-term outcomes.
Read also about hormonal replacement therapy

Supportive & Symptomatic Care
Alongside disease-modifying treatments, supportive care addresses daily functional challenges.
This includes physiotherapy to maintain mobility, respiratory management to support airway function, and cardiac monitoring to detect early complications. Hearing and vision support may also be required as the disease progresses.
Emerging Treatments
Research is increasingly focused on addressing the root genetic defect.
Gene therapy aims to introduce functional copies of the defective gene, potentially restoring enzyme production at a cellular level. Early clinical studies suggest potential benefits, though long-term outcomes are still under evaluation.
Prognosis & Life Expectancy
Prognosis depends heavily on the timing of intervention. Without treatment, the disease progresses rapidly, leading to significant morbidity and reduced life expectancy.
Early diagnosis and intervention can alter this trajectory. Patients receiving timely stem cell transplantation and ongoing care often experience improved survival and functional outcomes.
Read also about stem cell applications
Complications of Hurlers Syndrome
As the disease progresses, complications become more pronounced and interconnected.
Cardiac complications may arise due to valve thickening, while respiratory issues often stem from airway obstruction. Skeletal abnormalities can limit mobility, and neurological decline impacts cognitive function.
These complications require continuous monitoring and adaptive management strategies.
Living with Hurlers Syndrome
Living with Hurlers syndrome involves long-term coordination between medical care and daily support systems.
Patients often require multidisciplinary care, including pediatric specialists, neurologists, cardiologists, and rehabilitation teams. Beyond clinical management, family support plays a central role in maintaining quality of life.
Education, mobility assistance, and routine adaptations are essential components of long-term care.
Shortcomings of Current Hurlers Syndrome Therapies
Despite advancements, current treatments have limitations.
- Limited ability to reverse neurological damage
- Dependence on early diagnosis for effectiveness
- High treatment costs
- Accessibility challenges across regions
These gaps highlight the need for continued innovation.
Future of Hurlers Syndrome Gene Therapy
Gene therapy represents a shift from symptom management to potential disease correction.
Instead of supplementing the missing enzyme, this approach targets the genetic mutation itself. By restoring enzyme production at the source, it offers the possibility of long-term disease control.
Ongoing studies are evaluating safety, durability, and scalability of these therapies. While still in development, they represent one of the most promising directions in rare disease treatment.

Conclusion
Hurlers syndrome is a complex, multisystem disorder that requires early diagnosis and comprehensive care. While current treatments have improved outcomes, they remain limited in scope. Emerging gene therapy approaches offer a new direction, focusing on correcting the underlying cause rather than managing symptoms alone. Continued research and coordinated care will be central to improving long-term outcomes.
Explore more evidence-led healthcare insights and perspectives: MDForLives
Frequently Asked Questions
Is Hurlers syndrome genetic?
Yes. It is inherited in an autosomal recessive pattern caused by mutations in the IDUA gene.
What is the life expectancy of someone with Hurler’s syndrome?
Life expectancy varies, but early treatment significantly improves survival and quality of life.
What is the Hurler syndrome triad?
It typically includes developmental delay, skeletal abnormalities, and organ enlargement.
Can Hurler syndrome be detected before birth?
Yes. Prenatal genetic testing can identify the condition in at-risk pregnancies.
What rehabilitation strategies help Hurler patients?
Physical therapy, respiratory care, and multidisciplinary rehabilitation support functional ability over time.

MDForLives is a global healthcare intelligence platform where real-world perspectives are transformed into validated insights. We bring together diverse healthcare experiences to discover, share, and shape the future of healthcare through data-backed understanding.