Alpha 1 antitrypsin deficiency is a genetic condition that can affect both the lungs and liver, often remaining undiagnosed until symptoms appear. This hereditary disorder reduces the levels of alpha-1 antitrypsin (AAT), a protein that protects tissues from damage.
Without adequate protection, individuals may develop lung diseases such as COPD or liver complications over time. Early diagnosis and targeted treatment can significantly improve long-term outcomes.
What is Alpha 1 Antitrypsin Deficiency Autoimmune Disease?
Alpha 1 antitrypsin deficiency is not an autoimmune disease, but a genetic disorder caused by mutations in the SERPINA1 gene.
This gene controls production of alpha-1 antitrypsin, a protein made in the liver that protects the lungs from inflammation caused by enzymes such as neutrophil elastase.
When AAT levels are low or dysfunctional:
- lung tissue becomes vulnerable to damage
- abnormal protein may accumulate in the liver
- both respiratory and liver diseases can develop
Symptoms of Alpha-1 Antitrypsin Deficiency
Symptoms vary depending on whether the lungs or liver are affected.
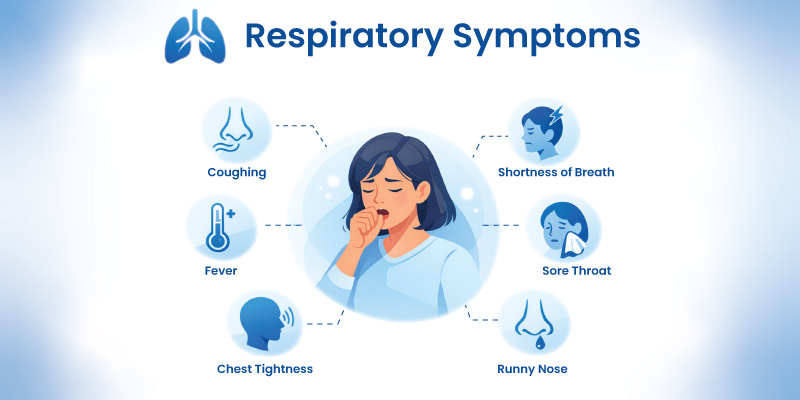
Lung-Related Symptoms
- shortness of breath
- wheezing
- chronic cough
- reduced exercise tolerance
- early onset COPD
Liver-Related Symptoms
- fatigue
- jaundice
- abdominal swelling
- unexplained liver disease
Symptoms may appear earlier than typical COPD, often between ages 20 and 50.
Causes of Alpha-1 Antitrypsin Deficiency
The condition is caused by inherited mutations in the SERPINA1 gene.
These mutations lead to:
- reduced production of AAT
- production of abnormal AAT proteins
- accumulation of defective proteins in the liver
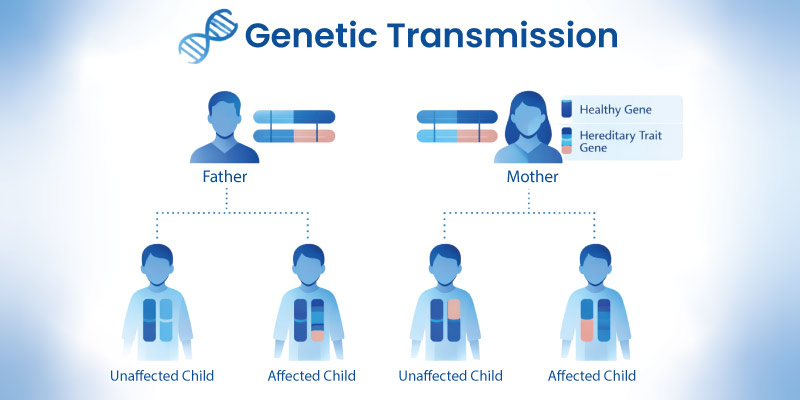
Alpha-1 Antitrypsin Deficiency Inheritance
It follows an autosomal codominant inheritance pattern.
This means:
- each parent contributes one copy of the gene
- severity depends on gene variants inherited
- individuals may be carriers or affected
Family history plays a key role in risk assessment.
Risk Factors
Certain factors increase the likelihood of disease progression:
- smoking (major risk factor for lung damage)
- environmental pollutants
- family history of AAT deficiency
- occupational exposure to dust or chemicals
Avoiding smoking is the most critical preventive measure.
Complications of Alpha-1 Antitrypsin Deficiency
Untreated AAT deficiency can lead to:
- chronic obstructive pulmonary disease (COPD)
- emphysema at a young age
- liver cirrhosis
- liver failure
- increased risk of liver cancer
Early diagnosis helps reduce complication risk.
How Alpha-1 Antitrypsin Deficiency Is Diagnosed
Diagnosis involves a combination of clinical and laboratory evaluation.
Blood Tests
Measure levels of alpha-1 antitrypsin.
Genetic Testing
Identifies mutations in the SERPINA1 gene.
Lung Function Tests
Assess breathing capacity and airflow limitation.
Imaging
Chest CT scans may detect early lung damage.
Screening is especially important in patients with unexplained COPD.
Alpha-1 Antitrypsin Deficiency Treatment
Treatment focuses on managing symptoms and slowing disease progression.
Augmentation Therapy
Augmentation therapy involves intravenous infusion of AAT protein to increase protective levels in the blood.
This therapy is commonly used in patients with lung involvement.
Medications
- bronchodilators
- inhaled corticosteroids
- antibiotics for infections
These help manage respiratory symptoms.
Read also about Antibiotics and Gut Health.
Oxygen Therapy
Used in advanced lung disease to improve oxygen levels.
Liver Management
Severe liver disease may require monitoring or, in advanced cases, liver transplantation.
Lifestyle Changes for Management
Lifestyle plays a major role in disease progression.
Avoid Smoking
Smoking accelerates lung damage significantly.
Maintain Healthy Diet
Balanced nutrition supports liver and lung health.
Exercise Regularly
Improves lung capacity and overall fitness.
Avoid Environmental Triggers
Reduce exposure to dust, chemicals, and pollutants.
Foods to Avoid:
Certain dietary choices may help reduce liver strain:
- excessive alcohol
- high-fat processed foods
- sugary beverages
- foods contributing to inflammation
A liver-friendly diet supports long-term health.
What is the Life Expectancy of Someone with Alpha-1?
Life expectancy varies based on:
- early diagnosis
- smoking status
- severity of lung or liver involvement
- access to treatment
Individuals diagnosed early and managed properly can live near-normal lifespans.

When Should You Get Tested?
Testing is recommended for:
- individuals with early-onset COPD
- unexplained liver disease
- family history of AAT deficiency
- persistent respiratory symptoms without clear cause
Early detection allows timely intervention.
Alpha-1 Antitrypsin Deficiency in Children
In children, AAT deficiency primarily affects the liver.
Symptoms may include:
- prolonged jaundice
- poor growth
- liver enlargement
Some children improve over time, while others require long-term monitoring.
Living With Alpha-1 Antitrypsin Deficiency
Managing AAT deficiency requires ongoing care.
Key strategies include:
- regular medical follow-ups
- adherence to treatment plans
- vaccination against respiratory infections
- lifestyle adjustments
With proper management, many individuals maintain good quality of life.
Conclusion
Alpha-1 antitrypsin deficiency is a hereditary condition that can significantly impact lung and liver health if left undiagnosed. However, with early detection, appropriate treatment, and lifestyle adjustments, disease progression can be managed effectively.
Increasing awareness and screening among high-risk individuals remains critical to improving outcomes.
Frequently Asked Questions
When should COPD patients be screened for alpha-1 deficiency?
Patients diagnosed with COPD at a younger age or without a clear cause should be screened.
Should family members be tested for alpha-1 deficiency?
Yes. Because it is a genetic condition, family members may benefit from testing to identify carriers or affected individuals.
Does AAT deficiency get worse with age?
Disease progression depends on lifestyle factors, especially smoking. Proper management can slow progression significantly.
What are the first signs of alpha-1 antitrypsin deficiency?
Early signs often include shortness of breath, chronic cough, or unexplained liver issues.
What vaccinations are recommended for AATD patients?
Vaccinations for influenza, pneumonia, and hepatitis are commonly recommended to reduce complications.

MDForLives is a global healthcare intelligence platform where real-world perspectives are transformed into validated insights. We bring together diverse healthcare experiences to discover, share, and shape the future of healthcare through data-backed understanding.